 5 hours ago
1
5 hours ago
1

한국은 신약 허가에 걸리는 기간이 미국 등 다른 선진국에 비해 월등히 긴 것으로 나타났다. 국내 바이오회사는 신약 허가가 지체돼 해외 경쟁사와의 출시 경쟁에서 밀리고 투자금 회수에도 어려움을 겪고 있다.
14일 한국경제신문이 코스닥시장에 기술특례로 상장한 바이오기업 112곳의 2023년 1월부터 2025년 2월까지 신약 임상시험계획(IND) 신청 현황을 조사한 결과 IND 승인이 나오는 데 걸린 기간이 식품의약품안전처는 평균 128일(32건), 미국 식품의약국(FDA)은 29일(8건), 호주 인체연구윤리위원회(HREC)는 29일(7건)로 나타났다.
식약처는 IND를 신청한 바이오기업에 30일 안에 심사 결과를 통보하는 것이 원칙이다. 하지만 기한 내 승인을 내준 사례는 0건이었다. 최장 약 1년이 걸린 사례도 있었다. 백신 개발 전문기업 셀리드는 항암 면역치료 백신의 IND를 신청한 뒤 승인받기까지 337일 걸렸다. 같은 기간 FDA는 한국 바이오기업이 신청한 IND를 모두 30일 이내에 처리했고, HREC는 한 건만 30일을 넘겼다. FDA와 HERC 역시 30일 안에 IND를 심사하도록 돼 있다.
신약을 출시하기 위해서는 1상부터 3상까지의 임상시험 과정을 거쳐야 한다. 임상시험 단계별로 보건당국에 IND를 제출해 승인받아야 한다. 이 때문에 임상시험 허가가 늦어지면 전반적인 신약 개발 일정에 차질이 빚어진다.
업계 관계자는 “바이오기업은 허가가 늦어지면 ‘갑 중의 갑’인 식약처에 항의조차 하지 못하고 기약 없이 기다릴 수밖에 없다”고 토로했다.
30일 내 심사결과 통보가 원칙, 실제 유명무실…최장 1년 걸려
식약처 "자료 부족해 보완 요청"…업계 "불필요한 자료로 시간 허비"
세계에서 가장 빨리 디지털 의료기기 분야 신제품을 개발한 한국 A사는 출시 경쟁에서 밀렸다. 식품의약품안전처의 임상시험 심사 과정이 지체된 여파다. 규정에 따르면 식약처는 30일 내에 임상시험계획(IND)을 심사해 업체에 알려야 한다. 식약처는 1년이 넘도록 심사를 끌었고, 그 사이에 미국 경쟁사가 ‘최초 타이틀’을 가져갔다.
◇유명무실 ‘30일 내 승인’ 기한
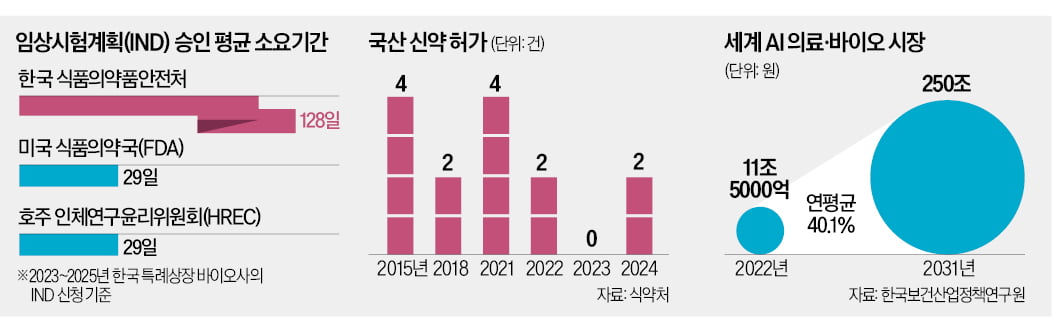
14일 한국경제신문이 특례상장 바이오기업 112곳의 2023년 1월부터 2025년 2월까지 임상 1~3상 IND 신청 현황을 조사한 결과 승인이 나오는 데 식약처는 평균 128일(32건), FDA는 29일(8건), 호주 인체연구윤리위원회(HREC)는 29일(7건)이 걸렸다.
IND 변경에 대한 승인 역시 제대로 지켜지지 않고 있다. IND 변경은 투약 일정, 임상시험 시행 기관과 환자 추가 모집 등 기존 IND 승인을 받아 진행하던 임상에서 변경 사항이 있으면 신청한다. 식약처는 16건 중 한 건을 제외하고 모두 기한을 넘겨 변경 승인을 내줬다. FDA는 한국 바이오기업이 신청한 7건의 IND 승인과 변경 모두 100% 30일 내 규정을 지켰다. HREC에 변경 승인을 신청한 사례는 없었다.
◇줄 잇는 자료 보완 요구에 하세월
국내 특례상장 바이오회사는 대부분 ‘계열 내 최초’ 또는 ‘계열 내 최고’ 신약을 개발 중이다. 세계 시장에서 경쟁 약물 대비 개발 속도가 빠르거나 아무도 하지 않는 기술로 개발한 신약이어야만 기술수출이 가능하기 때문이다.
하지만 업계는 디지털 의료기기, 유전자가위, 세포치료제 등 차세대 바이오 기술은 식약처 심사가 더 늦어진다고 토로한다. CJ바이오사이언스는 2022년 말과 2023년 초에 마이크로바이옴(장내 미생물) 면역항암제 IND를 FDA와 식약처에 각각 신청했다. IND 승인이 나오기까지 FDA는 27일, 식약처는 76일이 걸렸다.
업계는 식약처의 늑장 심사가 차세대 기술에 대한 ‘이해 부족’ 때문이라고 지적한다. 한 바이오기업 관계자는 “최첨단 기술이라도 FDA는 이미 정립된 가이드라인만 따라가면 되는 데 비해 식약처는 어떤 자료를 심사해야 하는지 모르기 때문에 방대한 자료를 수개월에 걸쳐 요구한다”며 “최신 기술은 업체에 규제를 알아서 마련해 오라고 요구하기도 한다”고 했다. 식약처 관계자는 “IND 신청 검토 과정에서 자료가 부족하거나 미비하면 신청인에게 보완 자료를 요청한다”며 “신청인으로부터 보완 자료를 받는 데 시간이 걸리거나, 제조시설 실태조사가 필요한 경우 등은 승인 심사 기간이 추가로 소요된다”고 말했다.
◇갈 길 먼 규제과학, 한국 걸음마 단계
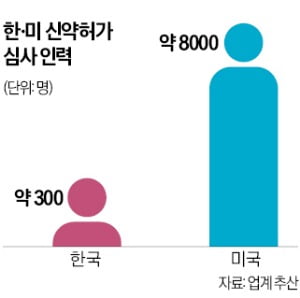
전문가들은 식약처가 차세대 기술 심사를 제대로 하기 위해서는 ‘규제과학’에 투자해야 한다고 입을 모은다. 의약품 안전성·유효성·품질 등의 평가 기술을 개발하고 심사 인력도 확충해야 한다는 것이다.
FDA는 규제과학 관련 별도 조직인 규제과학혁신센터(CERSI)와 광범위연구기관공고(BAA) 프로그램을 통해 꾸준히 투자하고 있다. 2010년 설립한 CERSI는 대학 연구진이 신약의 안전성, 효능, 품질을 평가하기 위한 새로운 도구와 기준, 접근 방식을 개발한다. 미국 존스홉킨스대 스탠퍼드대 예일대 등 유수 대학이 참여하고 있다. 2012년부터 운영한 BAA는 바이오회사, 학술기관 등을 포함해 광범위한 외부 기관 참여를 장려한다. FDA는 2024년에만 2450만달러를 투입했다.
식약처는 2021년부터 규제과학에 예산을 배정하기 시작했다. 식약처가 최수진 국민의힘 의원에게 제출한 자료에 따르면 규제과학 예산은 2021년 31억원, 2022년 50억원, 2023년 74억원, 2024년 191억원에 불과하다. 업계 관계자는 “코로나19 팬데믹 극복에 큰 역할을 한 메신저리보핵산(mRNA) 백신 개발 기간을 통상 10년에서 11개월로 단축할 수 있었던 건 FDA의 규제과학의 힘”이라며 “포스트 팬데믹에 대비하기 위해서라도 규제과학에 투자해야 한다”고 말했다.
김유림 기자 youforest@hankyung.com











 English (US) ·
English (US) · 